
Scientists take a step toward understanding 'jumping genes' effect on the genome
Using state-of-the-art statistical methods, a team of researchers said they may have taken a leap closer to understanding a class of jumping genes, sequences that move to different locations in the human genome, which is the body’s complete set of DNA. They added that the work could lead to insights into the evolution of the human genome, as well as have implications for several diseases, including cancer.
The sequences, referred to as Long Interspersed Elements-1 — often called LINE-1 or L1s — constitute a class of transposable elements, also known as "jumping genes" because they can move throughout the genome, said Di Chen, doctoral student in the Intercollege Graduate Program in Genetics. He said that geneticists and medical researchers are keen to understand these jumping genes and their interaction with the genome for a number of reasons.
"First, it's the only active LINE family that's still jumping around in our genome,” said Chen. "The human genome consists of different sequences and this particular retrotransposon, L1, makes up over 17% of it. Structure-wise it's very important, too. It gives rise to many other transposable element sequences and, throughout the course of evolution, it expanded the size of our genome. It also plays an important role in the regulation of its functions."
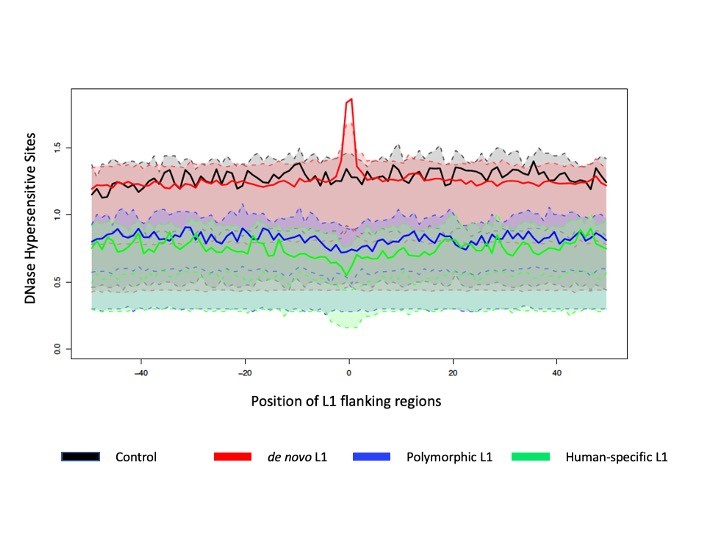
The researchers, who report their findings in a recent issue of Molecular Biology and Evolution, were able to build integrative models of L1s’ evolution, including where L1s tend to integrate and where they tend to remain — or become fixed — in the genome, according to Kateryna Makova, professor of biology, Verne M. Willaman Chair of Life Science, and director of the Center for Medical Genomics, Huck Institutes of the Life Sciences, Penn State.
"What we found out is quite interesting," said Makova, who is also an associate of the Institute for Computational and Data Sciences(ICDS). "This transposable element integrates and ends up remaining in very different genomic landscapes. Usually, they integrate mainly into particular parts of the genome, but then, after some evolutionary time, they end up being retained in other genomic locations."
The researchers also found that, although the integration and fixation processes are influenced by many different genomic features, L1s are more than just bits of DNA bouncing through the genome; they actually affect the genomic landscape surrounding them.
“There were a few studies before that hinted at this. Our study suggests that this is true and it’s something that is quite novel — the L1s influence the evolution of the genome,” said Makova.
The study may also lead to models that will give researchers a deeper understanding of L1s.
“By building models, as we have done, we could have a better idea of the role played by L1s in diseases and other important traits that are encoded in the genome,” said Makova. “For example, depending on where the L1s land as they move around, we could find whether they have a strong effect, a minor effect, or no effect at all. Of course, any of these predictions would need to be validated in computational and experimental studies, but it’s important that we are getting close to being able to do that.”
Although this work is preliminary, the insights also might lead to practical and clinical applications because the study shows that L1s are not passive entities, as once thought, but are active players in the genome. Some L1 insertions are also thought to cause interference with protein coding within the genome.
“It’s important now to take this research towards medical applications,” said Makova. “First of all, similar analysis can be done based on the datasets of L1 integrations in cancer, but also in other diseases. It’s known that some L1s play important roles in neurological diseases. For example, brain DNA sequencing studies are finding polymorphic L1 insertions. We could use the same methodology and apply it to more clinically relevant datasets.”
The team analyzed three genome-wide datasets of human L1s representing newly integrated, polymorphic (present in some individuals but absent in others), and human-specific L1s, together with 49 genomic landscape features collated from other studies.
They used a class of advanced statistical methods, called functional data analysis, to build models and precisely analyze the genomic landscape where L1s integrate and become fixed. The methods offered a way to analyze these genomic landscapes at high resolution, according to Marzia A. Cremona, assistant professor of operations and decision systems, Université Laval, and adjunct assistant professor of statistics, Penn State.
“We tested for differences in genomic landscape features, between regions where we had L1s and regions where we didn’t have these elements,” said Cremona. “Then we took the most important features to build a model to help us understand how all of these features work together.”
Compared to other methods, functional data analysis techniques are better at exploiting high-resolution data, thus offering researchers a richer view of the interplay between L1s and the genomic landscape,” said Francesca Chiaromonte, professor of statistics at Penn State, Lloyd and Dorothy Foehr Huck Chair in Statistics, and director of the Genome Sciences Institute, one of the Huck Institutes of the Life Sciences. "For example, we used these techniques to compare features among regions where L1s inserted a long time ago, a short time ago, or a very recent time ago with areas devoid of L1s."
“Using functional data analysis, instead of contrasting average values, we can contrast curves; that is, the shapes of measurements observed at very high resolution,” said Chiaromonte, who also is an ICDS affiliate. “Observing not just differences in averages, but differences in shapes of curves, allows us to better pinpoint relationships and effects.”
Robi D. Mitra, Alvin Goldfarb Distinguished Professor of Computational Biology and professor at Washington University School of Medicine, and his student Zongtai Qi also worked with the team.
The Penn State Clinical and Translational Sciences Institute, Penn State Institute for Computational and Data Sciences, and Eberly College of Science supported this work. Additional support was provided under grants from the Pennsylvania Department of Health using Tobacco Settlement and CURE Funds.